
Bab 2: Docking Molekuler
Memahami dasar-dasar, aplikasi, serta kelebihan dan kekurangan metode docking molekuler
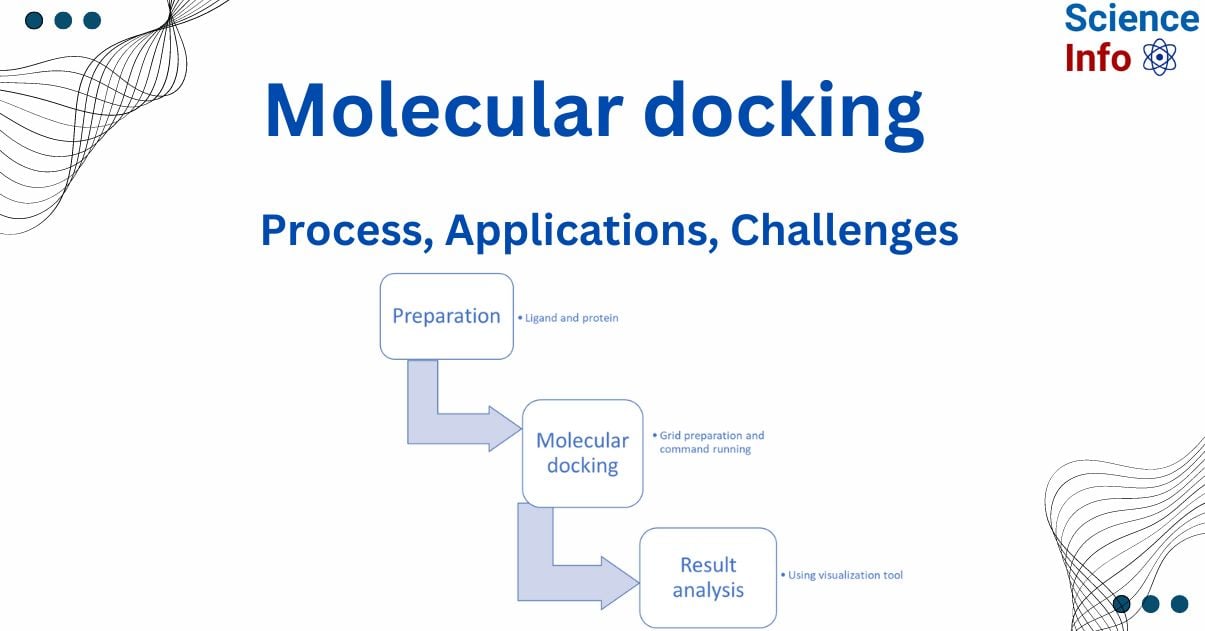
3 Key Takeaways
- Definisi : Docking molekuler adalah teknik komputasional untuk memprediksi interaksi antara molekul, terutama interaksi antara ligand dan protein.
- Parameter : Proses docking mengevaluasi energi ikatan, jenis ikatan (seperti ikatan hidrogen, elektrostatik, van der Waals, dan hidrofobik), serta konformasi dan orientasi optimal ligand.
- Aplikasi dan Tantangan: Banyak perangkat lunak tersedia untuk simulasi docking, namun metode ini memiliki kelebihan dalam efisiensi dan prediksi awal serta keterbatasan terkait akurasi dan fleksibilitas protein.
Pengertian Docking Molekuler
Docking molekuler adalah suatu metode dalam bioinformatika dan biologi molekuler yang digunakan untuk memprediksi interaksi antara dua molekul, terutama interaksi antara molekul obat (ligand) dengan target biologis seperti protein, enzim, DNA, maupun molekul biokimia lainnya. Teknik ini bekerja melalui simulasi komputer untuk menentukan orientasi optimal ligand ketika berikatan dengan suatu protein.
Konsep dasar dari docking molekuler sering kali diibaratkan dengan analogi "gembok dan kunci" atau "sarung tangan" yang menggambarkan bagaimana ligand (sebagai kunci) masuk ke situs aktif protein (sebagai gembok) untuk membentuk kompleks yang stabil. Tujuan utama dari proses docking adalah untuk memprediksi konformasi dengan energi ikatan yang paling rendah, sehingga menandakan interaksi yang paling stabil dan kemungkinan besar terjadi dalam kondisi biologis nyata.
Aspek yang Dinilai pada Docking Molekuler
Interaksi Antara Ligand dengan Protein
Dalam proses docking molekuler, evaluasi dilakukan dengan melihat interaksi spesifik yang terjadi antara ligand dan protein. Pemeriksaan ini melibatkan beberapa parameter kunci yang dapat memberikan gambaran komprehensif mengenai kualitas dan kekuatan interaksi.
Energi Ikatan
Energi ikatan atau energi bebas pengikatan (binding free energy) merupakan parameter utama yang digunakan untuk mengukur kekuatan interaksi antara ligand dan protein. Nilai energi yang lebih negatif menunjukkan ikatan yang lebih kuat dan lebih stabil. Energi ikatan dihitung dengan mempertimbangkan kontribusi gaya-gaya yang terlibat, seperti:
- Gaya van der Waals: Interaksi non-kovalen yang berperan dalam stabilisasi posisi ligand dalam situs aktif.
- Interaksi elektrostatik: Tarikan antara muatan positif dan negatif yang membantu orientasi ikatan.
- Ikatan hidrogen: Bentuk interaksi spesifik antara donor dan akseptor yang sangat penting dalam stabilisasi kompleks.
- Interaksi hidrofobik: Kontak antara bagian nonpolar pada ligand dan protein yang meningkatkan affinity pada lingkungan non-air.
Jenis-jenis Ikatan
Berbagai jenis ikatan dianalisis untuk menentukan seberapa baik ligand berinteraksi dengan situs aktif protein. Jenis-jenis ikatan tersebut meliputi:
- Ikatan Hidrogen: Ikatan ini sangat penting untuk pembentukan kompleks karena kestabilan dan spesifikasinya.
- Ikatan Elektrostatik: Tarikan antara muatan-muatan pada ligand dan protein yang membantu mengarahkan posisi pengikatan.
- Ikatan van der Waals: Meskipun relatif lemah, interaksi ini memberikan kontribusi total yang signifikan terutama pada kontak atom yang dekat.
- Interaksi Hidrofobik: Interaksi yang terjadi ketika bagian nonpolar berinteraksi untuk menghindari kontak dengan air, sehingga meningkatkan stabilitas kompleks dalam larutan berair.
Konformasi dan Orientasi Ligand
Selain energi ikatan dan jenis ikatan, proses docking juga menilai konformasi spatial dan orientasi ligand terhadap situs pengikatan pada protein. Beberapa poin utama yang dievaluasi adalah:
- Posisi Optimal: Posisi di mana ligand dapat memasuki situs aktif dengan orientasi yang memungkinkan interaksi paling efektif.
- Fleksibilitas Molekul: Baik ligand maupun protein dapat mengalami perubahan konformasi untuk mencapai fit terbaik. Analisis ini penting karena bidang fleksibilitas kedua molekul dapat mempengaruhi hasil akhir interaksi.
- Adaptasi Struktur: Perubahan konformasi yang terjadi selama proses pengikatan juga memberikan wawasan mengenai dinamika interaksi molekuler.
Analisis Kontak Residual
Selain menghitung nilai energi, analisis mendalam juga dilakukan untuk memeriksa interaksi antara asam amino tertentu pada protein dan bagian-bagian ligand. Evaluasi kontak residu membantu mengidentifikasi area kunci yang terlibat dalam pengikatan, sehingga dapat memandu optimasi desain obat.
Aplikasi Docking Molekuler
Seiring dengan pesatnya perkembangan komputasi dan algoritma, berbagai perangkat lunak telah dikembangkan untuk keperluan docking molekuler. Aplikasi-aplikasi ini bervariasi dari yang sederhana hingga yang kompleks, dan masing-masing memiliki keunggulan serta algoritma tersendiri yang disesuaikan dengan kebutuhan riset. Berikut adalah beberapa aplikasi populer yang sering digunakan:
Perangkat Lunak Docking Populer
- AutoDock dan AutoDock Vina: Merupakan salah satu perangkat lunak paling populer. AutoDock Vina khususnya dikenal karena kecepatan dan akurasinya dalam menghitung energi afinitas serta menemukan konformasi optimal ligand terhadap protein. Kedua perangkat lunak ini sangat banyak digunakan dalam penelitian desain obat dan virtual screening.
- DOCK: Salah satu paket perangkat lunak awal yang digunakan untuk docking molekuler. Meski dianggap klasik, DOCK masih digunakan terutama dalam studi screening farmakofor dan pencocokan bentuk antara ligand dengan situs aktif.
- GOLD (Genetic Optimization for Ligand Docking): GOLD memanfaatkan algoritma genetika untuk mengeksplorasi ruang konformasi dari ligand dan memberikan kemampuan tangani fleksibilitas baik pada ligand maupun protein secara lebih optimal, meskipun perangkat lunak ini bersifat komersial.
- Glide (bagian dari Schrodinger Suite): Glide menggunakan pencarian grid-based untuk menghasilkan hasil docking dengan cepat dan akurat. Dengan fungsi scoring yang komprehensif, Glide banyak diaplikasikan dalam pengembangan dan optimasi molekul obat.
- Aplikasi Pendukung Lainnya: Terdapat pula perangkat lunak seperti PyRx, PatchDock, serta alat bantu visualisasi seperti PyMOL, LigPlot+, dan Biovia Discovery Studio yang mendukung proses dari tahap persiapan hingga analisis interaksi hasil docking.
Integrasi dengan Metode Lain
Hasil dari simulasi docking molekuler sering kali digunakan sebagai dasar awal untuk melakukan simulasi dinamika molekuler (Molecular Dynamics) yang dapat menguji kestabilan interaksi secara lebih dinamis di lingkungan yang menyerupai kondisi in vivo. Metode ini juga dapat dipadukan dengan analisis kuantum untuk verifikasi lebih mendalam dari mekanisme interaksi.
Contoh Aplikasi pada Penelitian Eksperimental
Beberapa studi telah menerapkan docking molekuler untuk mengidentifikasi kandidat penghambat enzim atau protein target dalam penyusunan desain obat baru. Misalnya, penelitian yang memanfaatkan ekstrak tanaman untuk menghambat enzim ACE2 melalui simulasi docking telah menunjukkan bahwa sejumlah senyawa memenuhi kriteria molekuler tertentu yang dapat mengganggu interaksi kritis dengan protein target. Hal ini membuka jalan untuk penelitian lanjutan yang menggabungkan simulasi komputer dengan eksperimen in vitro dan in vivo.
Kelebihan dan Kekurangan Docking Molekuler
Kelebihan Docking Molekuler
Docking molekuler memiliki beberapa keunggulan yang membuatnya sangat berharga di dunia riset farmasetik dan biologi molekuler:
-
Efisiensi Waktu dan Biaya: Metode komputasional memungkinkan analisis interaksi ribuan senyawa dalam waktu yang relatif singkat tanpa perlunya eksperimen laboratorium yang mahal dan memakan waktu. Pendekatan ini sangat membantu dalam tahapan awal screening kandidat obat.
-
Prediksi Interaksi Molekuler: Memberikan wawasan mendalam mengenai interaksi spesifik antara ligand dan protein, sehingga memungkinkan perancangan dan optimasi molekul obat secara terarah serta pengurangan risiko kegagalan pada tahap eksperimen in vitro dan in vivo.
-
Fleksibilitas Modeling: Beberapa perangkat lunak mendukung simulasi fleksibel pada kedua komponen, sehingga dapat menangani pergerakan dan adaptasi konformasi secara lebih realistis pada saat interaksi berlangsung.
-
Integrasi dengan Analisis Lain: Hasil docking molekuler dapat dikombinasikan dengan simulasi dinamika molekuler, perhitungan kuantum, dan analisis ADME (absorbsi, distribusi, metabolisme, dan ekskresi) sehingga menghasilkan validasi yang lebih komprehensif terhadap potensi senyawa.
Kekurangan Docking Molekuler
Meskipun semakin banyak digunakan dan disempurnakan, metode docking molekuler juga memiliki beberapa keterbatasan yang perlu diperhatikan:
- Keterbatasan Akurasi: Hasil prediksi dari docking molekuler sangat bergantung pada kualitas struktur 3D ligand dan protein. Jika data input tidak akurat, maka hasil yang diperoleh juga dapat menyimpang dan tidak mencerminkan interaksi nyata.
- Fleksibilitas Protein yang Terbatas: Banyak metode docking menganggap protein sebagai struktur yang relatif kaku, sehingga tidak sepenuhnya mampu mengakomodasi perubahan konformasi yang terjadi secara alami. Hal ini dapat menyebabkan prediksi false positive atau false negative.
- Skoring Function yang Tidak Sempurna: Fungsi penilaian (scoring function) yang digunakan untuk menghitung energi ikatan terkadang tidak mampu menampung semua faktor kompleks yang mempengaruhi interaksi molekuler, seperti efek lingkungan larutan dan interaksi multikomponen.
- Kompleksitas Sistem Biologis: Walaupun simulasi docking memberikan gambaran awal, sifat statis dari model komputasional tidak selalu mampu mensimulasikan dinamika dan kompleksitas interaksi di dalam sistem biologis nyata, sehingga membutuhkan validasi eksperimental lebih lanjut.
Contoh Tabel Perbandingan Perangkat Lunak Docking
Berikut adalah tabel yang membandingkan beberapa perangkat lunak untuk docking molekuler berdasarkan parameter utama seperti metode scoring, kecepatan, dan kemampuan menangani fleksibilitas:
| Perangkat Lunak | Metode Scoring | Kecepatan | Fleksibilitas Protein | Kemudahan Penggunaan |
|---|---|---|---|---|
| AutoDock / AutoDock Vina | Empirik, berbasis energi bebas | Tinggi | Terbatas (model rigid atau semi-flexible) | Mudah, banyak dokumentasi |
| DOCK | Screening farmakofor, analisis bentuk | Sedang | Terbatas | Menengah |
| GOLD | Algoritma genetika dan scoring khusus | Sedang hingga tinggi | Lebih fleksibel | Komersial, antarmuka ramah pengguna |
| Glide | Grid-based scoring function | Tinggi | Lebih optimal pada fleksibilitas | Profesional, bagian dari suite Schrodinger |
Kesimpulan
Docking molekuler telah menjadi salah satu metode terpenting di dalam desain dan pengembangan obat modern. Dengan memprediksi interaksi antara ligand dan protein, teknik ini tidak hanya menghemat biaya dan waktu, tetapi juga memberikan wawasan mekanistik yang mendalam yang membantu para peneliti dalam mengoptimalkan kandidat obat.
Walaupun terdapat keterbatasan, seperti akurasi prediksi yang sangat bergantung pada kualitas struktur 3D dan asumsi kekakuan protein, kelebihan dari efisiensi serta kemampuannya untuk mengintegrasikan data dari simulasi lebih lanjut menjadikan docking molekuler sebagai alat yang sangat berguna dalam riset farmasi dan biologi molekuler. Kombinasi dengan metode seperti dinamika molekuler atau analisis kuantum diharapkan dapat mengatasi kekurangan tersebut dan mendukung penelitian eksperimental lebih lanjut.
Secara keseluruhan, pemahaman mendalam mengenai parameter evaluasi – termasuk energi ikatan, jenis ikatan, orientasi ligand, serta kontak interaksi residu – sangat penting untuk menginterpretasikan hasil simulasi docking. Penggunaan berbagai aplikasi docking modern, mulai dari AutoDock Vina, DOCK, GOLD, hingga Glide, memungkinkan fleksibilitas dalam penelitian, namun setiap alat memiliki keunggulan dan kekurangan tersendiri yang harus dipertimbangkan sesuai dengan tujuan penelitian.
Referensi
https://www.wikiwand.com/id/articles/Docking_(molekuler)
https://id.wikipedia.org/wiki/Docking_(molekuler)
http://repositori.unsil.ac.id/9859/12/12.+BAB+II.pdf
https://p2k.stekom.ac.id/ensiklopedia/Docking_(molekuler)
https://stifera.ac.id/2023/04/15/desain-obat-masa-depan-menggali-potensi-molekuler-docking-untuk-menciptakan-obat-baru/
https://pmc.ncbi.nlm.nih.gov/articles/PMC5425816/
https://online-journal.unja.ac.id/jop/article/download/36116/18716/108454
https://www.researchgate.net/post/Molecular_Docking_using_PyRx_software
https://jurnal.ugm.ac.id/majalahfarmaseutik/article/download/59297/31297
https://biotek.fsm.undip.ac.id/v1/2022/06/28/pengenalan-molecular-docking-dan-aplikasinya/
https://ojs.unida.ac.id/Agrohalal/article/download/6753/3768
https://ffarmasi.uad.ac.id/pelatihan-in-silico-docking-molekuler/
https://pubmed.ncbi.nlm.nih.gov/28510083/
Last updated February 17, 2025